Comparative Genomics for Plant Pathologists
Introduction
Whole-genome sequencing (WGS) has fundamentally transformed bacterial plant pathology. What once required a combination of phenotypic characterization, pathogenicity testing, biochemical assays, and a limited set of molecular markers can now be achieved using a single high-quality genome sequence.
The rapid adoption of Illumina, Oxford Nanopore Technologies (ONT), PacBio sequencing and hybrid sequencing approaches has enabled laboratories to generate bacterial genomes routinely. However, sequencing alone is not sufficient. The true value of WGS lies in comparative genomics—the process of comparing genomes to understand species boundaries, evolutionary relationships, pathogen emergence, host adaptation, and outbreak dynamics.
For plant pathologists, comparative genomics provides answers to several essential questions:
- What species is this isolate?
- How is it related to previously characterized strains?
- Does it belong to a known epidemic lineage?
- Which genes may contribute to pathogenicity or host specificity?
- Can genomic evidence improve diagnostics?
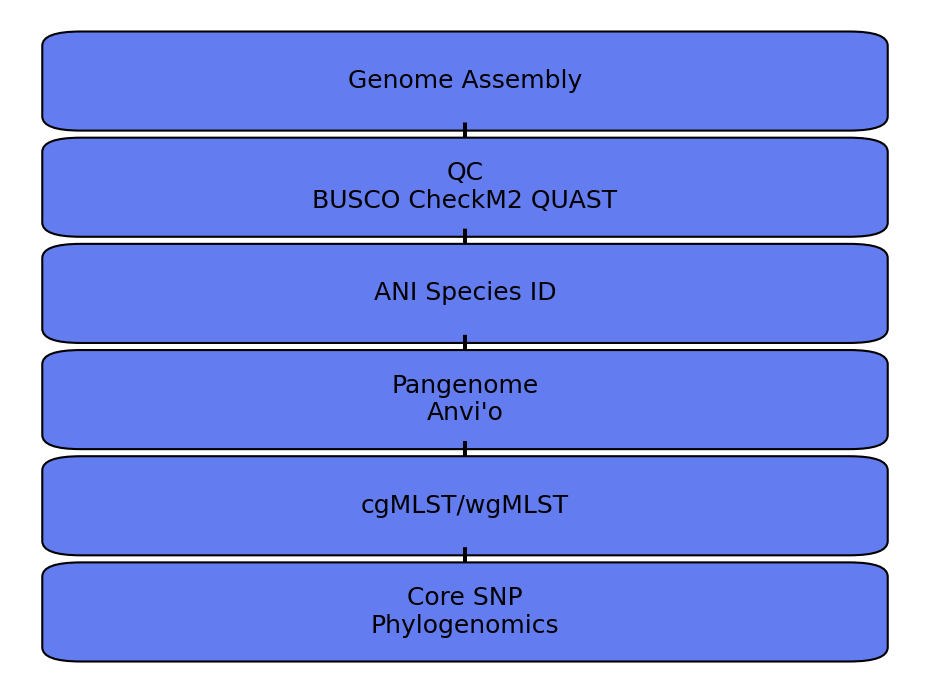
Modern bacterial genomics therefore represents a continuum of resolution, ranging from species identification through Average Nucleotide Identity (ANI) to strain-level characterization using core-genome SNP analysis. The choice of method depends entirely on the biological question being addressed.
Genome Quality: The Foundation of Comparative Genomics
Comparative genomic analyses are only as reliable as the genomes on which they are performed.
Errors introduced during DNA extraction, sequencing, assembly, contamination, or annotation can propagate through downstream analyses and result in incorrect conclusions. Consequently, genome quality assessment should always precede comparative analyses.
During the EURL Workshop 2025 on plant pathogen genomics, we emphasized the need for standardized genome quality criteria before entering taxonomic or phylogenetic workflows.
Recommended Quality Thresholds
| Metric | Recommended Threshold |
|---|---|
| Illumina quality | Q30 or higher |
| ONT quality | Q20 or higher |
| BUSCO completeness | >90%, ideally >97% |
| Contamination | <5% |
| N50 | >100 kb preferred |
| Number of contigs | <200 |
| ANI confirmation | ≥95% |
| Genome size | Within 10% of expected |
| GC content | Consistent with species |
BUSCO has become one of the most widely adopted tools for assessing assembly completeness because it evaluates the presence of conserved single-copy orthologs expected within a lineage rather than relying solely on assembly statistics.
Similarly, CheckM and CheckM2 estimate contamination and completeness based on lineage-specific marker genes, thereby identifying mixed cultures and problematic assemblies before comparative analyses are performed. Although CheckM2 is faster, CheckM estimates are still better and comparable with BUSCO genome completeness and contamination.
Comparative Genomics as a Hierarchical Framework
One of the most important principles in bacterial genomics is that no single method provides optimal resolution for every question.
Instead, comparative genomics should be viewed as a hierarchy.
Species-Level Resolution
At the broadest level, researchers seek to determine species identity.
This is typically achieved using:
- Average Nucleotide Identity (ANI)
- GTDB classification using skANI or GTDBTk
- GenomeRxiv taxonomic assignment (GTDB/NCBI)
- Marker gene phylogenies
Population-Level Resolution
Once species identity has been established, researchers often investigate:
- Host-associated lineages
- Pathovars
- Sequevars
- Clonal complexes
At this level, methods such as pangenomics, cgMLST, and wgMLST become informative.
Strain-Level Resolution
For epidemiological investigations, outbreak tracing, and transmission studies, maximum resolution is required.
Core-genome SNP analyses frequently provide the highest discriminatory power available for bacterial isolates.
Species Identification with Average Nucleotide Identity
Average Nucleotide Identity (ANI) is now regarded as the genomic gold standard for bacterial species delineation.
ANI quantifies nucleotide similarity across homologous genomic regions shared between two genomes. Large-scale genomic analyses have demonstrated that bacterial strains belonging to the same species generally exhibit approximately 95–96% ANI, making ANI a practical replacement for traditional DNA-DNA hybridization.
Comparative Genomics for Bacterial Plant Pathogens
Species Identification Tools
Several software packages are available for average nucleotide identity (ANI)-based species identification:
- skANI
- pyANI-plus
- FastANI
- GTDB-Tk
- GenomeRxiv
In routine workflows, skANI has proven particularly useful because it combines speed with the ability to screen large databases such as GTDB. Identification typically takes only seconds per genome when using pre-built sketch databases.
GenomeRxiv is another valuable web-based resource with an API (using the BrookLIN CLI) that provides taxonomic assignment based on GTDB and NCBI databases. It first uses a fast mash-based approach to identify the closest reference genome, followed by ANI calculation for species confirmation.
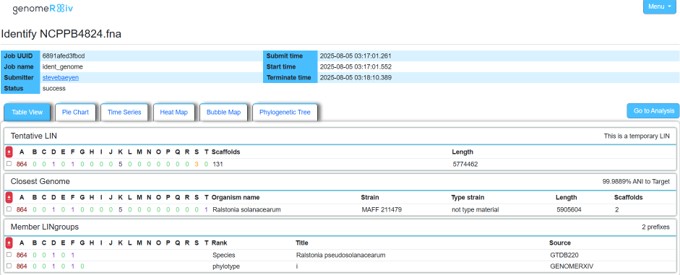
Practical Example — Routine Diagnostics
Assembled genome
↓
skANI / GTDB / GenomeRxiv
↓
ANI ≥ 95%
↓
Species confirmed
Beyond Species: Why ANI Is Not Enough
Although ANI is highly effective for species identification, it rapidly loses discriminatory power among closely related strains. For example:
- RSSC sequevars frequently exceed 98–99% ANI.
- Closely related Pseudomonas syringae populations often exhibit extreme genomic similarity.
- Epidemiologically linked outbreak strains may differ by only a handful of mutations.
In such situations, researchers require higher-resolution approaches — this is where pangenomics and population genomics become essential.
Understanding the Bacterial Pangenome
The concept of the pangenome was introduced to describe the entire gene repertoire of a species. A pangenome can be divided into three components:
Core Genome
Genes present in nearly all isolates, including those involved in:
- DNA replication
- Transcription
- Translation
- Central metabolism
Accessory Genome
Genes present only in subsets of isolates, frequently including:
- Virulence factors
- Secretion systems
- Resistance determinants
- Mobile genetic elements
Unique Genome
Genes found in individual strains, often resulting from:
- Horizontal gene transfer
- Plasmid acquisition
- Prophage integration
- Niche-specific adaptation
Comparative pangenomic studies across thousands of microbial genomes have demonstrated that accessory genomes contribute substantially to ecological specialization and pathogen evolution. For plant pathogens, accessory genes can often explain important differences in host range, aggressiveness, and environmental adaptation.
Pangenome Analysis Using Anvi’o
Among the available pangenome platforms, Anvi’o has emerged as one of the most powerful and flexible solutions. Anvi’o integrates:
- Functional annotation
- Gene clustering
- ANI calculations
- Phylogenomics
- Interactive visualization
- Comparative analyses
Typical Anvi’o Workflow
Genome assemblies
↓
Gene annotation
↓
Gene clustering
↓
Core/accessory identification
↓
Phylogenomic analysis
The platform enables simultaneous investigation of genomic diversity, evolutionary history, and functional gene content — particularly valuable for population studies involving Ralstonia, Pseudomonas, Xanthomonas, Dickeya, and related plant pathogens.
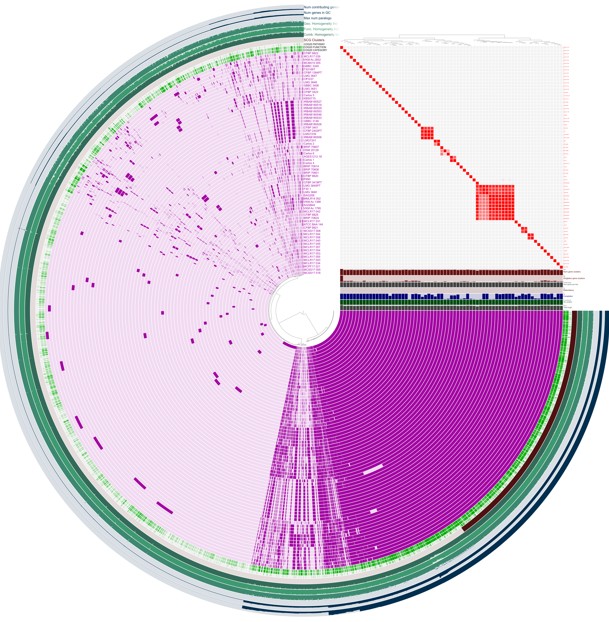
Gene-by-Gene Comparative Genomics
An alternative to SNP-based analyses is gene-by-gene typing, which compares allelic variants across predefined loci rather than individual nucleotide positions.
Core Genome MLST (cgMLST)
Focuses on genes shared by nearly all isolates. Benefits include:
- Robust standardization
- Easy inter-laboratory comparison
- Reproducible nomenclature
Whole Genome MLST (wgMLST)
Extends analyses to accessory loci, increasing discriminatory power and facilitating outbreak investigations.
One widely adopted platform is chewBBACA, which supports:
- Schema development
- Allele calling
- Core genome extraction
- Population analyses
These methods provide a practical compromise between ANI and SNP-level analyses.
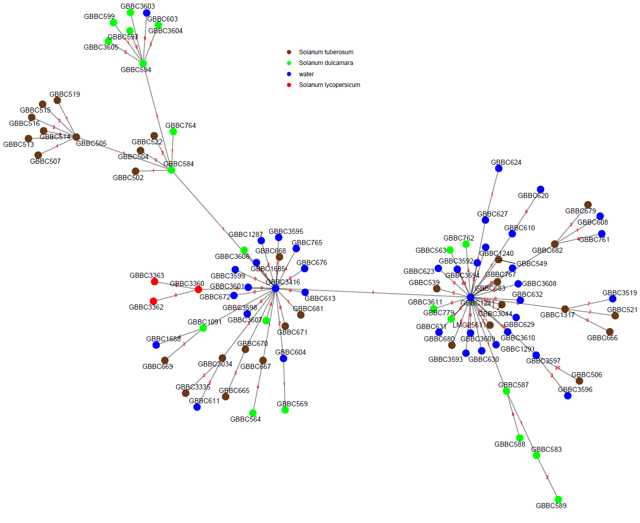
Core Genome SNP Analysis
When the highest possible resolution is required, SNP-based approaches remain the preferred strategy. Rather than comparing genes, SNP approaches compare individual nucleotide substitutions across the core genome.
Applications include:
- Outbreak investigations
- Epidemiological studies
- Clonal lineage tracking
- Evolutionary analyses
Typical Workflow
Reference selection
↓
Read mapping
↓
Variant calling
↓
Variant filtering
↓
Recombination filtering
↓
Phylogenetic reconstruction
A recent addition to my variant calling workflow is:
- using a pangenome reference for mapping, which improves SNP detection in highly diverse populations using the SuperPang package.
- using a consensus variant caller like VariantDetective, that uses state of the art variant callers like FreeBayes, Clair3 and GATK HaplotypeCaller to generate a high-quality consensus variant call set.
The Importance of Recombination Filtering
Many bacterial species undergo homologous recombination. Without accounting for recombination, phylogenetic reconstructions may incorrectly interpret horizontally transferred DNA as evolutionary divergence.
Gubbins was specifically developed to address this problem by detecting genomic regions containing unusually high densities of SNPs indicative of recombination and removing them from downstream phylogenetic analyses. Benchmarking studies demonstrated that recombination-aware analyses substantially improve bacterial phylogenetic reconstruction.
Practical SNP Phylogenomics
A common workflow combines:
- Snippy
- Gubbins
- IQ-TREE2
- iTOL
Workflow
Illumina reads
↓
Snippy mapping
↓
Core SNP alignment
↓
Gubbins recombination filtering
↓
IQ-TREE phylogeny
↓
iTOL visualization
This approach remains one of the most robust strategies for strain-level investigations in bacterial plant pathology.
Choosing the Right Genomic Resolution
The optimal methodology depends on the biological question:
| Biological Question | Recommended Approach |
|---|---|
| What species is this? | ANI |
| Is it correctly classified? | ANI + GTDB |
| Which lineage does it belong to? | Pangenome analysis |
| Is it related to other populations? | cgMLST |
| Is it part of an outbreak? | wgMLST |
| Are isolates nearly identical? | Core SNPs |
| Which genes explain adaptation? | Pangenomics |
No single method should be viewed as universally superior. Instead, complementary genomic approaches provide progressively finer resolution and biological insight.
Applications in Plant Pathology
Comparative genomics now supports numerous applications across plant health research:
- Diagnostics — Genome-informed diagnostics improve assay design, specificity evaluation, and marker selection. Unique genome signatures between target and non-target genomes can be found using the SkIf2 package for qPCR assay detection.
- Taxonomy — ANI and phylogenomics increasingly replace classical taxonomic frameworks.
- Epidemiology — SNP analyses enable high-resolution tracing of pathogen introductions and dissemination events.
- Population Genomics — Pangenomics and MLST approaches reveal population structure and host adaptation.
- Virulence Research — Accessory genome analyses facilitate the identification of genes associated with pathogenicity and host specificity.
Lessons Learned
After analyzing hundreds of bacterial plant pathogen genomes, several observations repeatedly emerge:
- Most comparative genomics problems begin with genome quality issues.
- ANI is invaluable for species identification but insufficient for strain discrimination.
- Accessory genomes frequently contain biologically important traits.
- Recombination must often be considered explicitly.
- Long-read sequencing significantly improves assembly contiguity and plasmid recovery.
- Reproducible workflows are more important than constantly switching software.
Ultimately, comparative genomics should be viewed as an integrated framework rather than a single analytical method.
Conclusion
Comparative genomics has become a central pillar of modern bacterial plant pathology. High-quality genome assemblies now enable rapid species identification, population analyses, phylogenomic reconstruction, and investigation of adaptive gene content.
By integrating ANI, pangenomics, MLST approaches, and core-genome SNP analyses, researchers can move seamlessly from species classification to outbreak investigation and evolutionary biology.
As sequencing technologies continue to improve and genomic databases expand, the future challenge is no longer generating genomes. The challenge is converting genomic data into biological understanding.
References
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212.
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications. 2018.
- Hyun JC, Monk JM, Palsson BO. Comparative pangenomics: analysis of microbial pathogen pangenomes reveals conserved global structures of genetic and functional diversity. BMC Genomics. 2022.
- Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Research. 2015.
- EURL Workshop 2025 – WGS in Practice: Building Reliable Genomes for Plant Pathogen Diagnostics and Research. Steve Baeyen.
Enjoy Reading This Article?
Here are some more articles you might like to read next: