Illumina vs. Nanopore for Plant Pathogens — A Practical Overview
Why this comparison matters in plant pathology
High‑throughput sequencing has become a routine tool in plant pathogen research, diagnostics, and surveillance. A lot of labs are now asking: should we use Illumina or Nanopore sequencing for our plant pathogen projects? The answer isn’t one‑size‑fits‑all—it depends on your specific goals, resources, and the biology of your pathogens. For bacterial plant pathogens such as Pseudomonas, Xanthomonas, Ralstonia, Erwinia, and related taxa, the choice between Illumina short‑read sequencing and Oxford Nanopore long‑read sequencing has direct consequences for:
- turnaround time
- assembly quality
- plasmid detection
- downstream phylogenetics and diagnostics
This post provides a practical, lab‑oriented comparison of Illumina and Nanopore sequencing, grounded in real Snakemake workflows used for plant‑pathogen genomics.
The technologies in one sentence
- Illumina → short, highly accurate reads; ideal for SNPs, surveillance, and diagnostics
- Nanopore → long, real‑time reads; ideal for complete genomes and plasmids

Read characteristics and assembly implications
Illumina (short reads)
- Typical reads: 2×150 bp
- Very low per‑base error rate
- Assemblies are accurate but often fragmented
Best suited for:
- routine diagnostics
- SNP‑based phylogenetics
- cgMLST / MLST
- high‑throughput surveillance
Nanopore (long reads)
- Read lengths: 5 kb – 100 kb+
- Higher raw error rate (improving rapidly)
- Assemblies often closed
Best suited for:
- reference genome generation
- plasmid and genomic island detection
- structural variation
- rapid outbreak investigation

Diagnostics vs discovery
| Use case | Illumina | Nanopore |
|---|---|---|
| Routine diagnostics | ✅ | ⚠️ |
| SNP resolution | ✅ | ⚠️ |
| Complete genomes | ❌ | ✅ |
| Plasmid detection | ❌ | ✅ |
| Turnaround time | ⏳ | ⚡ |
In practice, both technologies complement each other rather than compete.
Snakemake workflows used in practice
The real impact of sequencing technology is best understood through the analysis workflows that follow.
Illumina bacterial assembly + QC workflow
Designed for high‑throughput Illumina paired‑end data, this pipeline performs:
- read QC and trimming
- de novo assembly
- assembly QC (QUAST, BUSCO, CheckM2)
- taxonomic validation
- unified HTML/PDF reporting
📦 Repository
https://gitlab.ilvo.be/genomics/wgs/illumina-bacterial-assembly-snakemake

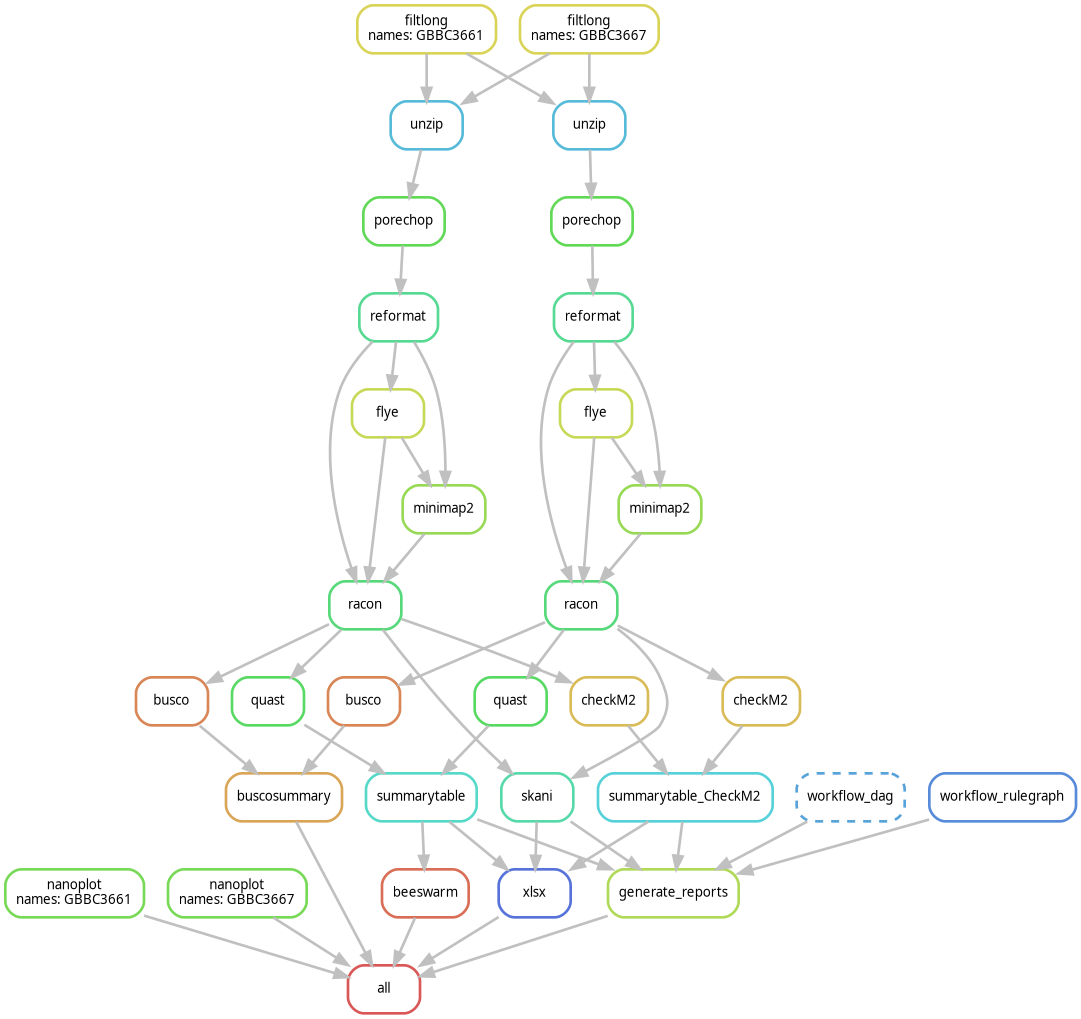
Nanopore‑only bacterial assembly workflow
Optimized for Oxford Nanopore R10.4.1 long‑read data, focusing on:
- long‑read filtering
- de novo assembly
- polishing and QC
- reference‑grade bacterial genomes
📦 Repository
https://gitlab.ilvo.be/stevebaeyen/nanopore_only_snakemake
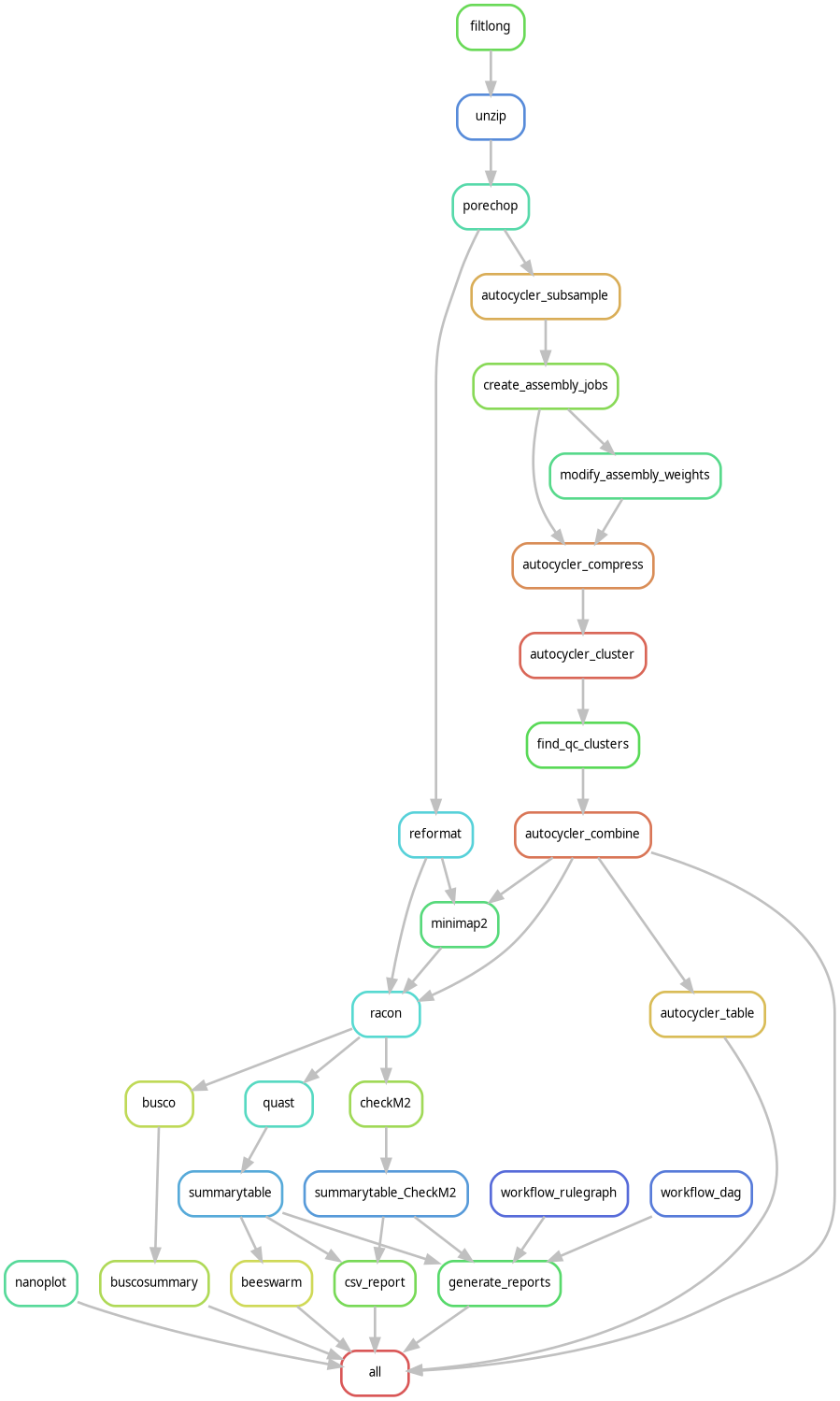
Nanopore Autocycler consensus workflow
For maximum assembly quality, this workflow integrates multiple assemblers through Autocycler and Snakemake.
Key features:
- read subsampling
- Flye / Canu / Plassembler consensus
- conflict resolution
- publication‑quality genomes
📦 Repository
https://gitlab.ilvo.be/stevebaeyen/nanopore_autocycler_snakemake
Assembly QC‑only workflow (technology‑agnostic)
Used when assemblies already exist (Illumina, Nanopore, or hybrid).
Includes:
- QUAST
- BUSCO
- CheckM2
- ANI‑based taxonomic checks
- Excel + HTML/PDF summaries
📦 Repository
https://gitlab.ilvo.be/stevebaeyen/bacterial-assembly-qc-snakemake

Decision tree: which pipeline should I use?
flowchart TD
A[Start: choose a pipeline] --> B{Do you have sequencing data?}
B -->|No| C{Main objective?}
C -->|Diagnostics or SNPs| D[Illumina sequencing and assembly pipeline]
C -->|Complete genome or plasmids| E[Nanopore sequencing and assembly pipeline]
B -->|Yes| F{Data type?}
F -->|Illumina reads| D
F -->|Nanopore reads| G{Assembly quality needed?}
F -->|Existing assemblies| H[Assembly QC pipeline]
G -->|Fast and good| I[Nanopore-only pipeline]
G -->|Reference-grade| J[Nanopore Autocycler pipeline]
Enjoy Reading This Article?
Here are some more articles you might like to read next: